




-
Project IDT2015-134
-
RFP Year2015
-
Awarded Amount$297,133DiseaseMalariaInterventionDrugDevelopment StageTarget ValidationCollaboration PartnersTakeda Pharmaceutical Company Limited , Medicines for Malaria Venture (MMV) , The University of Melbourne
Introduction and Background of the Project
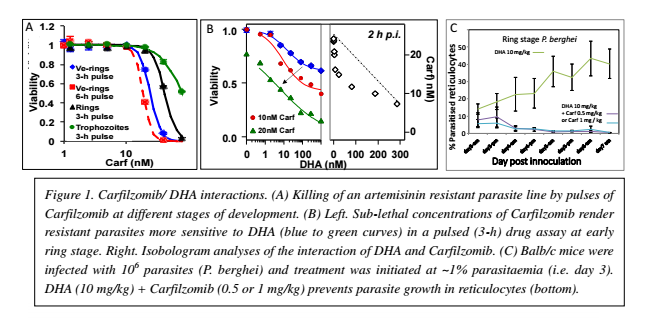
Plasmodium falciparum causes 200 million malaria infections and about 580,000 deaths each year (WHO World Malaria Report, 2014). Current malaria treatment is very heavily reliant on one class of drugs, known as the artemisinins. Thus it is extremely concerning that resistance to artemisinin has now arisen in Souteast Asia (Ashley et al., N Engl J Med 371, 411 (2014)). This leads to an urgent need for new antimalarials that can be used to overcome artemisinin resistance. In work leading to this project, we found that inhibitors of the plasmodium proteasome a proteinase complex that plays a critical role in degrading unfolded proteins - show parasiticidal activity against both artemisinin sensitive and resistant parasite - at all stages of intraerythroctyic development (see Figure 1, panel A). This includes the young (ring stage) form of the parasite, which is resistant to most other chemotherapeutic agents. Moreover we found that inhibitors of the proteasome strongly synergize artemisinin-mediated killing of P. falciparum in cultures of both sensitive and resistant strains (see Figure 1, panel B). Importantly, efficacy of the combination was also observed against P. berghei in a mouse model of malaria in vivo (see Figure 1, panel C). This indicates that a suitable proteasome inhibitor (i.e. with P. falciparum-specific activity) would be a promising lead in its own right and, would be particularly effective in combinations with artemisinins.
How can your partnership (project) address global health challenges?
Because malaria treatment is so heavily reliant on artemisinin combination therapies (ACTs), the spread of resistance to artemisinin will have very serious consequences. The World Health Organization has warned: "If resistance were to spread to – or emerge in – India or sub-Saharan Africa, the public health consequences could be dire. There is therefore a limited window of opportunity to avert a regional public health disaster, which could have severe global consequences." Thus it is extremely concerning that resistance is now evident in six Southeast Asian countries, including a region in Myanmar, close to the Indian border (Tun et al. Lancet Infect Dis 15, 415 (2015)). It has been estimated that even a 30% ACT failure rate worldwide would result in at least 116,000 extra deaths, and cost an additional US$385 million (AUD513M) in lost productivity each year (Lubell et al. Malar J 13, 452 (2014)). This project aims to develop assays that will enable the efficient identification of compounds that have highly specific and potent activity against the P. falciparum proteasome. These proteasome activity assays will form part of a test cascade that can be used to validate and characterize compounds from different sources.
What sort of innovation are you bringing in your project?
An exciting aspect of this project is that it brings together an academic partner (Tilley), a pharmaceutical industry expert (Dick) and a Product Development Partner (Burrows, MMV). We will build on the many years of work that have gone into developing proteasome inhibitors as anti-cancer agents; we will translate that knowledge to the development of new antimalarials. The enthusiastic involvement of Dr Dick is particularly important as he was involved in the discovery of Bortezomib and Ixazomib. He has an intense academic interest in the proteasome, particularly in targeting this enzyme for therapeutic purposes. A technical innovation of our project is the determination of a proteasome inhibitor "signature" that is associated with highly specific anti-plasmodium activity and resistance reversing activtiy. We anticipate that this chemical "signature" will comprise efficient interaction with both β2 and β5 subunits of the ART resistant P. falciparum proteasome, but weak interaction with the mammalian proteasome or interaction only with the β2 subunit of the mammalian proteasome. This aspect of the work will be important in the selection of drug candidates that will be both effective and safe for use in malaria patients.
Role and Responsibility of Each Partner
Through Dr. Larry Dick, Scientific Fellow, Oncology Clinical R&D, Millennium Pharmaceuticals, Inc. (a wholly-owned subsidiary of Takeda Pharmaceutical Company Limited) brings many years of experience in the development of proteasome inhibitors that can be translated to malaria. This includes lessons learned from the successful discovery, development and translation to clinic of bortezomib and ixazomib. Millenium Pharmaceuticals will host a 2-week visit from Dr. Stanley Xie, research scientist from the University of Melbourne. Stanley will work with Takeda scientists to learn relevant methods for studying human proteasome biology and biochemistry. During his visit, a Takeda library of proteasome inhibitors will be screened against the P. falciparum proteasome using the assays that Stanley has developed in the University of Melbourne laboratories.
Through, Dr. Jeremy Burrows, VP, Head of Drug Discovery, Medicines for Malaria Venture brings many years' experience in brokering collaborations for the development of antimalarial drugs. Dr Burrows is supporting the target validation and assay development work in the Project by providing expertise about test-bed systems. This will ensure that assays delivered are sufficiently robust and validated to support a test cascade for drug discovery. Dr. Burrows is also providing advice regarding future optimization phases in order to deliver Target Candidate Profiles suitable for the development of a preclinical candidate. He is also helping to broker access to Takeda compound libraries and providing advice on the compounds sets that emerge from this study.
The University of Melbourne is the designated development partner for this project and Professor Tilley is responsible for driving the project and working with Partners to develop the research plan, manage the project budget, ensure effective communication within the team and provide reports to GHIT. The University of Melbourne provides the laboratory space and personnel to develop assays that will enable the identification of compounds with highly specific and potent activity against the P. falciparum proteasome. This includes the development and validation of luminescence- and fluorescence-based activity assays employing Plasmodium extracts and purified proteasome preparations. These proteasome activity assays will form part of a test cascade to validate and characterize compounds from the Takeda libraries and other sources.
Final Report
1. Project objective
The Project aimed to establish assays that can be used to identify compounds with plasmodium-specific proteasome inhibitory activity, killing activity against Plasmodium falciparum cultures, and low mammalian cell toxicity. We also aimed to establish a working relationship with Takeda, Japan's largest pharmaceutical company, with a view to screening relevant Takeda libraries to identify a suitable proteasome inhibitor combination that can be translated to clinical use to overcome artemisinin resistance.
2. Project design
a) Purify stocks of 20S proteasome from P. falciparum cultures.
b) Generate stocks of recombinant mammalian PA28 (as a 20S activator).
c) Optimise a fluorogenic assay of the activity of Pf 20S proteasome for use in a compound screen.
d) Screen the purified Pf 20S proteasome against selected compounds from the Takeda proteasome inhibitor library.
d) Assess the activities of the compounds against the mammalian proteasome.
e) Determine the reversibility of binding of selected compounds from the above screens to the mammalian proteasome (as a measure of potential toxicity).
f) Determine substrates preferences for the human and Pf 20S proteasomes.
3. Results, lessons learned
a) Purified Pf 20S proteasome (~300 mg) was prepared from P. falciparum and was shown to be largely free of contamination with erythrocyte 20S proteasome.
b) Recombinant mammalian PA28 was generated (by Takeda Pharmaceuticals) and shown to be an efficient Pf20S activator.
c) The inhibitory activities of compounds from the Takeda proteasome inhibitor library were assessed against b1, b2 and b5 subunit activities of Pf 20S proteasome.
d) The inhibitory effects of the 131 compounds against both constitutive and immunoproteasome isoforms of human 20S were determined.
e) A proteasome-GLO assay (in mammalian Calu6 cells) was used to examine the reversibility of compounds selected from the above screens.
f) A library of ~6000 Ac-P3-P2-P1-AMC substrates was assessed as substrates for the human and Pf 20S proteasomes.
Differential substrate preferences were observed for Pf and human constitutive proteasomes. Compounds were identified that exhibit selectivity for the Pf20S proteasome b2 or potent inhibition of both b2 and b5. Compounds were identified that show little activity as inhibitors of human 20S proteasome in the proteasome-GLO assay but are potent inhibitors of parasite growth (and show some activity vs Pf20S). Compounds were identified that exhibit reversibility of action against human 20S proteasome and hence lower host toxicity.
The results suggest that it will be possible to design an inhibitor that is capable of co-inhibiting Pf proteasome b2 and b5 activities.
Aspartic acid is preferred by the Pf proteasome at the P1 position, while tryptophan is preferred at the P2 and P3 positions. This information will aid rational drug design.
We have established an excellent working relationship with Takeda Pharmaceutical, facilitated by placement of a research scientist from the Lead Institution into the Takeda premises for the compounds screen.
The rapid progress made enabled us to fast-track the compound library screening process.
Investment
Details
Portfolio
Advancing Portfolio
Proteasome inhibitors as new potent resistance-reversing antimalarials